goal: recent Emory HIV results and clustering consensus
- Emory HIV full genome sequencing shown at ASHG November 2012. - Follow up projects for stronger papers: - HIV genome from bulk mixtures and separated SGA samples. > Decovolute complex mixtures from clinical samples. - HIV GAG (1.5kb) sequencing. > Sequence 1 billion distinct genomes on a single chip ? - Clustering consensus analysis. - General framework: HIV, viral populations, 16S metagenomics, HLA diploid estimates, H.pylori cagY, BCR-ABL, cancer rare variant diagnostics. - Many components to be optimized. Next ================================HIV transmission biological problem:
- Examine full HIV genomes (9kb) from clincial transmission pairs. - Sequence complex mixtures of full HIV genomes (9kb) as well as single genomes physically separated by SGA. - PacBio Sequence 6 donor SGAs, 2 recipient SGAS, and pool of 60 bulk PCRs from donor. Try smartbells (overhang and blunt) and Tdt preps.
+-----------+------+---------------------------+
| sample | runs | comment |
+-----------+------+---------------------------+
| R463M.OH | 6 |new donor OH bulk |
| R463M.BL | 3 |new donor BL bulk |
| R463M.Td | 3 |new donor TdT bulk |
| R463F.sga | 2 |new recipient sga |
| R463M.sga | 6 |new donor sga |
| R880F.pool| 4 |old recipient pooled sga's |
| R880M.pool| 4 |old donor polled sga's |
+-----------+------+---------------------------+
- Best outcome: identify the exact HIV founder genome to the base that
transmitted between the donor and the recipient.
Next
================================
Clustering consensus framework for HIV full genome sequencing:
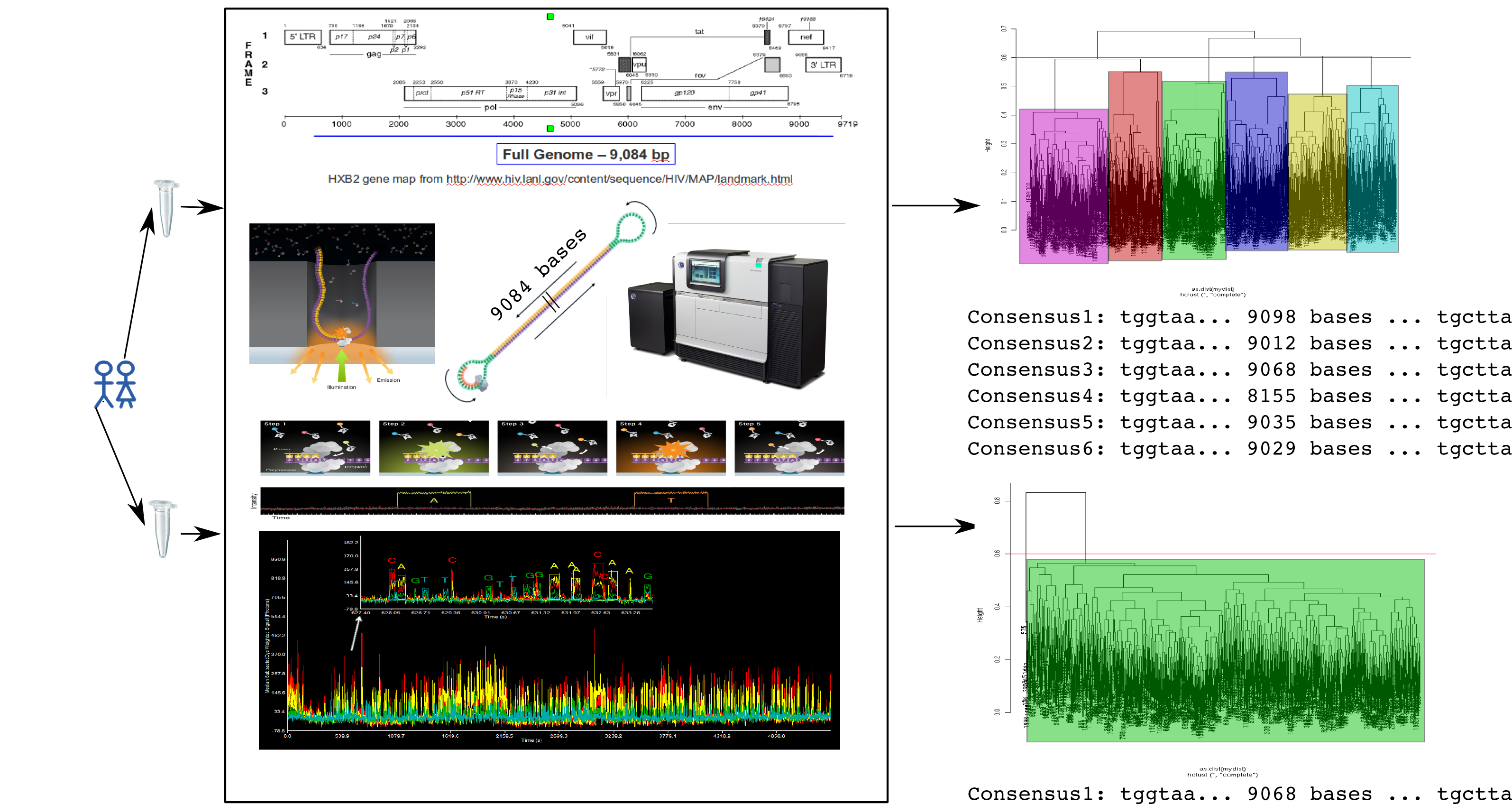 ----
The call:
python /home/UNIXHOME/mbrown/mbrown/workspace2012Q4/HIVSGASynMix/ConsensusClusterSubset.py \
--runDir cc-2450417-0020 \
--fasta run0020_s1_p0.fasta \
--ref HIVemory.fasta \
--spanThreshold=6400 \
--entropyThreshold=1.0 \
--basfofn 2450417-0020.bas.fofn \
> 2450417-0020.workflow.output 2>&1
--------
Workflow:
- estimate single best consensus using Quiver
- align all fully spanning reads to produce MSA
- feature select (rare) variant columns using entropy
- compute distance between all pairs of reads:
- Fraction of mismatches in MSA variant columns
- cluster all pairwise distances with agglomerative complete-linkage.
- stratify reads by cluster using threshold and recurse.
This is the "plan B" algorithm. Quiver is the
plan A that needs more time.
Next
================================
----
The call:
python /home/UNIXHOME/mbrown/mbrown/workspace2012Q4/HIVSGASynMix/ConsensusClusterSubset.py \
--runDir cc-2450417-0020 \
--fasta run0020_s1_p0.fasta \
--ref HIVemory.fasta \
--spanThreshold=6400 \
--entropyThreshold=1.0 \
--basfofn 2450417-0020.bas.fofn \
> 2450417-0020.workflow.output 2>&1
--------
Workflow:
- estimate single best consensus using Quiver
- align all fully spanning reads to produce MSA
- feature select (rare) variant columns using entropy
- compute distance between all pairs of reads:
- Fraction of mismatches in MSA variant columns
- cluster all pairwise distances with agglomerative complete-linkage.
- stratify reads by cluster using threshold and recurse.
This is the "plan B" algorithm. Quiver is the
plan A that needs more time.
Next
================================
HIV transmission clustering results:
- For 28 runs show the clustering diagrams.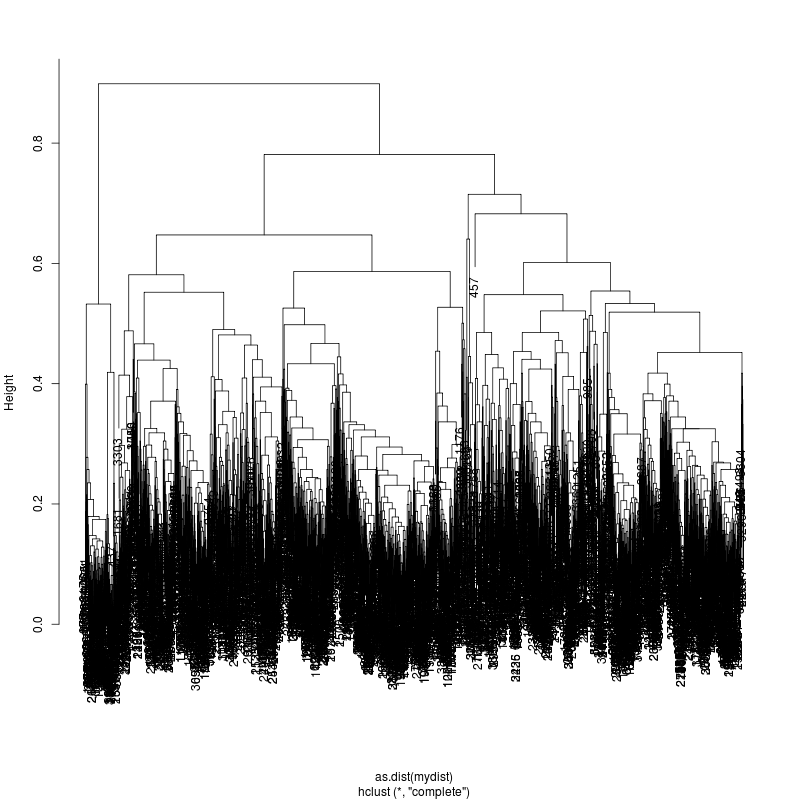
+-----------+------+---------------------------+
| sample | runs | comment |
+-----------+------+---------------------------+
| R463M.OH | 6 |new donor OH bulk |
| R463M.BL | 3 |new donor BL bulk |
| R463M.Td | 3 |new donor TdT bulk |
| R463F.sga | 2 |new recipient sga |
| R463M.sga | 6 |new donor sga |
| R880F.pool| 4 |old recipient pooled sga's |
| R880M.pool| 4 |old donor polled sga's |
+-----------+------+---------------------------+
README_RESULT_clusterimages.html
README_RESULT_clusterimages_9k.html
- How do you determine number of groups and cut the cluster ?
- Given the simple distance, use binomials to threshold noise:
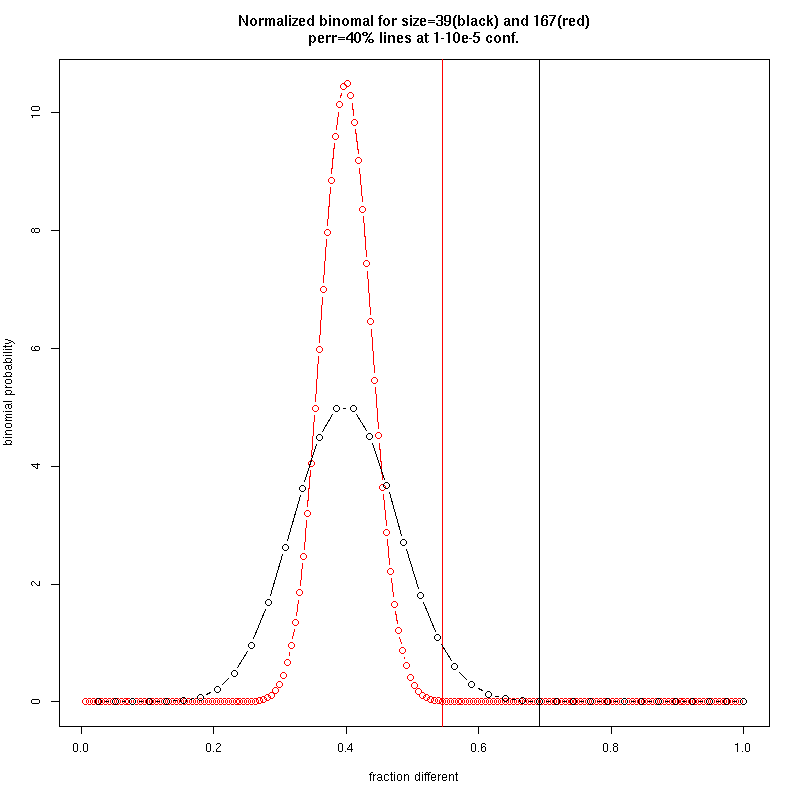
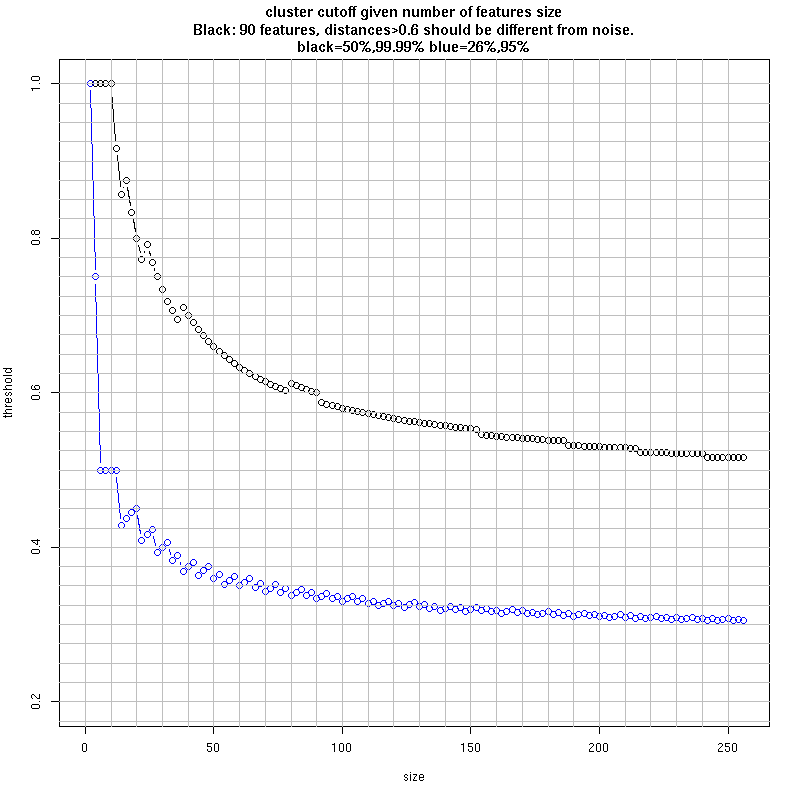 - Approximate statistical cutoff given identified variant positions:
- Approximate statistical cutoff given identified variant positions:
+-----------------+----------+------------+
| Run | Variants |Threshold |
+-----------------+----------+------------+
| POOLED bulk PCRs| ~150 |0.6 |
| SGA F runs | ~27 |0.725 |
| SGA M runs | ~20 |0.925 |
| 880F | ~3 |1.0 |
| 880M | ~130 |0.55 |
+-----------------+----------+------------+
- Problem SGAs are different from previous 21 SGAs:
README_RESULT_allClusters.html
Next
================================
Alignment of all Quiver SGA and bulks.
- How are single Quiver consensus estimates related disregarding subspecies? - Alignment of all 28 Quiver HIV estimates: allSGA.aln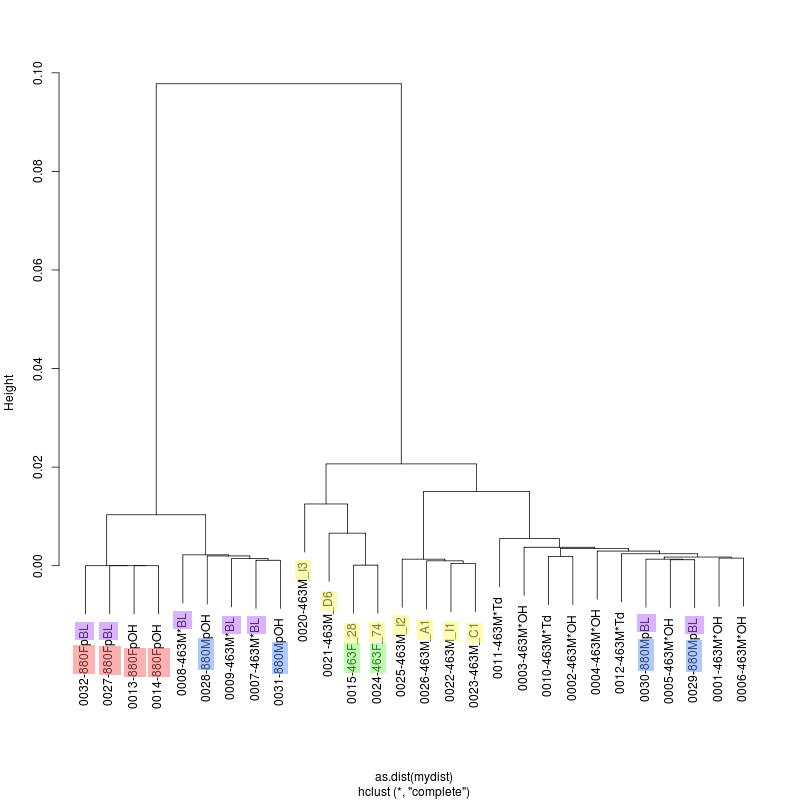 - Weird grouping of R463M*BL with R800MpOH and R463M*OH with R880MpBL.
- R463M_D6 is the closest to R463F, the founder virus? Present as a
subspecies in the bulk?
Next
================================
- Weird grouping of R463M*BL with R800MpOH and R463M*OH with R880MpBL.
- R463M_D6 is the closest to R463F, the founder virus? Present as a
subspecies in the bulk?
Next
================================