NIAID Close HIV Genomes
- GOAL: Sequence ~20x ~9kb unlabeled arbitrary HIV genomes that can differ by a single base or 1000's of bases on a single PacBio chip. - Samples to develop and test newly developed methods:
S#1: NL4.3 wild type (WT)
S#2: NL4.3 + K65R (AAA to AGA)
S#3: NL4.3 + M41L (ATG to TTG)
S#4: NL4.3 + T215Y (ACC to TAC)
S#5: NL4.3 + Y181C (TAT to TGT) + G190A (GGA to GCA)
M1 S#1-20%+S2-20%+S3-20%+S4-20%+S5-20% "easy" uniform mix 20%
M3 S#1-80%+S2-05%+S3-05%+S4-05%+S5-05% "harder" mix 5%
M5 S#5-50%+S#1-50% uniform 2 SNP phasing
- RESULTS:
- All pure samples agree except for two base differences.
- M5 correctly phased
- M1 correctly phased to five genomes except for half of one variant missed
- M3 not correctly identified.
- FUTURE WORK:
- Confirm sequencing "errors"
- Higher coverage of new chemistry should resolve harder cases.
- Many algorithmic techniques to be explored
- Tackle 20-mix after in-silico experiments.
================================
Previously
- Previous techniques: + CluCon: Good for genomes differing by >30 positions. Fully phased, base accurate results given about 100 reads. Universal but weaker "model-free" technique. Good results on 3-clone mixtures down to 5%. + LAA: Designed for HLA, barcoded samples, diploid variants. More complex variants allowed but not designed for. Good results on mixtures of large internal deletion HIV. - GAP: <30 variant positions and >>2 genomes ================================CluCon Basis
- Stronger modeling of sequencing error using homopolymer basis functions (A,C,G,T runs of 1,2,3,4,5,...) allowing for fine-scale identification and phasing of arbitrary mixtures of variants. - Models P( A.6 -> AAGAATTAA ) and P( A.4+T.2+A.2 -> AAGAATTAA ) - Learn models by sequencing exactly known templates and counting in regions with back-off models for unseen contexts. Visualization - Used to identify variant regions by excess of observations. - Used to phase positions using linear system matrix: M(i,j) = P(H_j -> H_i) Obs = M * Truth -> Truth = M^{-1} * Obs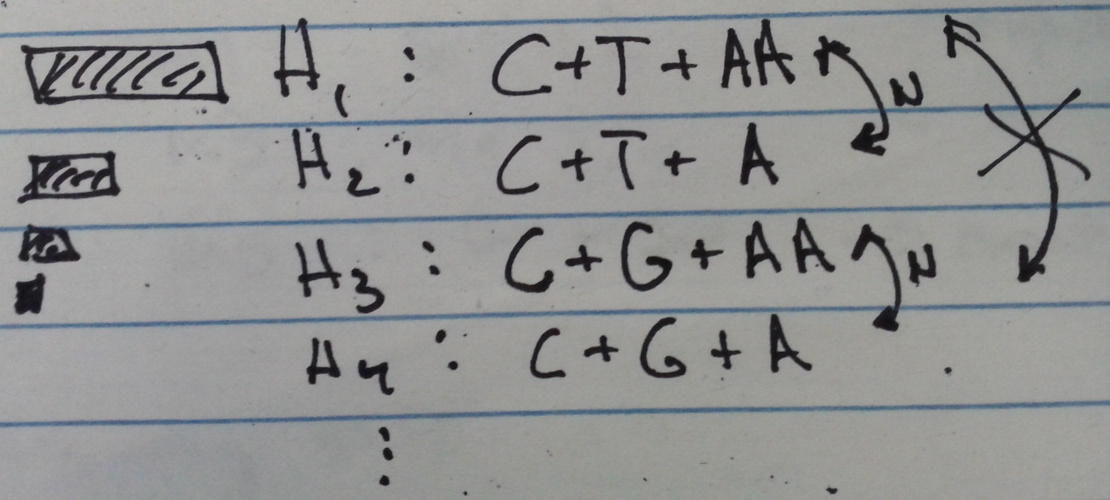 ================================
================================
Sequencing Results
- Good number of P4 full length genomes:
Sample #FL ConsensusLength
S1 2525 8911
S2 3036 8911
S3 2445 8911
S4 3526 8910
S5 2715 8910
M1 3853 8911
M3 2664 8911
M5 983 8911
- S1, M1, and M3 had the same consensus. M5 is equal to S5 rather than S1.
================================
Sequencing Results
- Sequencing is correct except two unaccounted errors:
msapos quiver HPbegin HPend context HPrange quiverbase truthbases type
ERROR! 6010 1201 6005 6025 ACA 6005 6025 C.A.3.T A ['A', 'A', 'A', '-', 'A'] delete
s#3 9850 1969 9835 9855 AAA 9835 9855 G.A.3.T A ['A', 'A', 'T', 'A', 'A'] mismatch
s#2 10215 2042 10205 10240 GAA 10205 10240 G.A.6.G A ['A', 'G', 'A', 'A', 'A'] mismatch
s#5 11955 2390 11950 11960 CTA 11950 11960 T.A.1.T A ['A', 'A', 'A', 'A', 'G'] mismatch
s#5 12090 2417 12080 12095 AGG 12080 12095 A.G.2.A G ['G', 'G', 'G', 'G', 'C'] mismatch
s#4 12441 2487 12435 12445 GGA 12435 12445 G.A.1.T . ['.', '.', '.', 't', '.'] insert
s#4 12441 2487 12440 12460 GGA 12440 12460 A.T.3.A . ['.', '.', '.', 't', '.'] insert
s#4 12465 2492 12460 12475 TAC 12460 12475 A.C.2.A C ['C', 'C', 'C', '-', 'C'] delete
ERROR! 28455 5690 28450 28460 GAT 28450 28460 A.T.1.G T ['T', 'T', 'T', 'T', '-'] delete
ERROR#1 in S#4: S#3
C....A....A....A....T G....A....A....A....T....G
C....-....A....A....T G....A....A....T....T....G
S#2
T....A....A....A....G....A....A....A....A....A....A....G
T....A....A....A....G....A....G....A....A....A....A....G
S#5
C....T....A....T....C + A....G....G....A....T
C....T....G....T....C A....G....C....A....T
S#4
T....G....G....G....G....A....T....T....T....A....C....C....A
T....G....G....G....G....At...T....T....T....A....-....C....A
Corrected:
T....G....G....G....G....A....T....T....T....A....C....C....A
T....G....G....G....G....A....T....T....T....T....A....C....A
ERROR#2 in S#5:
G....A....T....G....C
G....A....-....G....C
The two errors are upheld in other independent sequencing runs.
The observations suggest the consensus is correct with little
suggestion of the wild-type for the two "errors" (74/2309, 18/2023).
================================
Variant Positions
- All pure samples had no identified variant positions. - M5 identified 2 variant positions correctly - M1 identified 5 variant positions but should have 6 - M3 identified 0 variant positions but should have 6 ================================M5 Identified and Correctly Phased
[['G', 'A', 'A', '', 'G', '', 'AA', 'GG', 'G', 'G', 'A', 'TA', 'G', 'A', 'GT', 'AT', 'A', '', 'G'],
['C', 'G', '', 'G', 'CC', 'C', 'G', 'C', '', 'GC', 'GG', 'G', 'CA','A', 'C', 'G', 'C', '', 'TC']]
observed [359, 287, 51, 23, 21, 15, 12, 11, 11, 11, 10, 10, 9, 9, 7, 7, 6, 6, 6]
================================ top 1
'A+G+' [[0, 871], [287, 871]] 4.80251396237e-78
should add! 1 'A+G+'
================================ top 2
'A++' [[16, 871], [51, 871]] 2.783535288e-05 # 7% discounted minor = 51/(359+287+51)
'+G+' [[15, 871], [23, 871]] 0.250924327934
'G+CC+' [[12, 871], [21, 871]] 0.159007424968
'+C+' [[20, 871], [15, 871]] 0.495312158218
'AA+G+' [[11, 871], [12, 871]] 1.0
'GG+C+' [[12, 871], [11, 871]] 1.0
'G++' [[18, 871], [11, 871]] 0.261083327465
'G+GC+' [[4, 871], [11, 871]] 0.116946397511
'A+GG+' [[9, 871], [10, 871]] 1.0
'TA+G+' [[3, 871], [10, 871]] 0.0911245759195
'G+CA+' [[4, 871], [9, 871]] 0.265075360206
'A+A+' [[1, 871], [9, 871]] 0.0211469672287
'GT+C+' [[4, 871], [7, 871]] 0.547542762848
'AT+G+' [[3, 871], [7, 871]] 0.342354228124
'A+C+' [[3, 871], [6, 871]] 0.506690410552
'++' [[2, 871], [6, 871]] 0.287945566371
'G+TC+' [[4, 871], [6, 871]] 0.753202724374
*** converged! the following haplotypes and fractions explain all the data
0 'G+C+' 0.55572755418
1 'A+G+' 0.44427244582
================================
M1 Mostly Identified and Correctly Phased
- Should find six variant positions but only find five
Top p-values:
1660 11950 11960 T.A.1.T 3.682654e-131 # s#5
1682 12080 12095 A.G.2.A 1.148554e-119 # s#5
1371 9835 9855 G.A.3.T 5.278450e-108 # s#3
1411 10205 10240 G.A.6.G 9.658415e-92 # s#2
1734 12435 12445 G.A.1.T 5.180785e-91 # s#4 ? t-insert, where is the c-delete ?
...
1737 12460 12475 A.C.2.A 4.036372e-32 # S#4 c-delete
- The S#4 variant misses the c-delete
S#4
T....G....G....G....G....A....T....T....T....A....C....C....A
T....G....G....G....G....At...T....T....T....A....-....C....A
Corrected:
T....G....G....G....G....A....T....T....T....A....C....C....A
T....G....G....G....G....A....T....T....T....T....A....C....A
- The C-deletion is there but the p-value doesn't ring
it out.
id observed expected expectedProb over 3537.0
CC 2238.0 2434.0 0.688117608683
C 724.0 434.0 0.122587252492
CCC 165.0 133.0 0.0376396169989
....
- TODO: If I take 12435:12475, it is probably more significant because
it takes the two "bled" variant positions and puts them together.
- The phasing is correct ... just lacking the corresponding c-delete
================================ top 1
'AAT+AAAAAA+A+GG+A+' [[1, 1481], [119, 1481]] 1.12602313786e-33
should add! 1 'AAT+AAAAAA+A+GG+A+'
================================ top 2
'AAA+AGAAAA+A+GG+A+' [[0, 1481], [116, 1481]] 1.10841149061e-34
should add! 2 'AAA+AGAAAA+A+GG+A+'
================================ top 3
'AAA+AAAAAA+G+GC+A+' [[0, 1481], [114, 1481]] 4.13977487445e-34
should add! 3 'AAA+AAAAAA+G+GC+A+'
================================ top 4
'AAA+AAAAA+A+GG+A+' [[32, 1481], [65, 1481]] 0.00126598134044
'AA+AAAAAA+A+GG+A+' [[16, 1481], [59, 1481]] 5.37653495232e-07
'AAA+AAAAAA+A+GG+AT+' [[0, 1481], [55, 1481]] 5.22373769102e-17
should add! 6 'AAA+AAAAAA+A+GG+AT+'
...
*** converged! the following haplotypes and fractions explain all the data
0 'AAA+AAAAAA+A+GG+A+' 0.331125827815
1 'AAT+AAAAAA+A+GG+A+' 0.19701986755
2 'AAA+AGAAAA+A+GG+A+' 0.192052980132
3 'AAA+AAAAAA+G+GC+A+' 0.188741721854
6 'AAA+AAAAAA+A+GG+AT+' 0.091059602649
- If I stratify to the 'AAA+AAAAAA+A+GG+AT+' variant then the correct
single C-deletion is observed 27 times, so stratification might
resolve exactly.
================================
M3 Is Missed!
- P-values of the correct variant positions
V1 V2 V3 V4 target
1371 9835 9855 G.A.3.T 2.799849e-14 9840
1411 10205 10240 G.A.6.G 1.138599e-19 10210
1660 11950 11960 T.A.1.T 5.627766e-05 11955
1682 12080 12095 A.G.2.A 1.138794e-06 12085
1734 12435 12445 G.A.1.T 5.464434e-30 12440
1737 12460 12475 A.C.2.A 2.831993e-04 12465
- If I threshold at these values I get a minimum of 80 false
positions with no hope of phasing.
- The data do support the variants but is underpowered to detect it:
S#5 A->G
11950 11960 T.A.1.T {
'T....A....T': 1888,
'T....-....T': 103,
'-....A....T': 99,
'T....A....-': 86,
'Ta...A....T': 84,
'T....G....T': 44, **
'T....At...T': 39,
'Tt...A....T': 31,
'T....Aa...T': 28,
'Tg...A....T': 23,
'T....Ag...T': 17,
'A....A....T': 15,
'-....A....-': 15,
'T....-....-': 15,
'T....Ac...T': 15,
'Tc...A....T': 13,
'T....T....T': 13,
================================