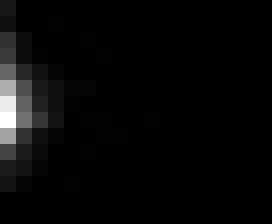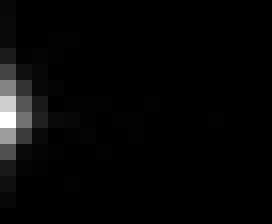PacBio Error Plots: Base + Homopolymer length
Plots showing the number of correct identity bases versus number of
incorrect identity bases for every base and homopolymer length.
This is based on sequencing a single HIV genome. I look at alignments
regions of the form: x+y^n+z where (x!=y and y!=z)
I then count the number of correct bases (y) versus the number of
incorrect bases (!y). For all regions, I sum the counts and plot in a
matrix that has 14 rows and 17 columns. The rows represent 0:13
correct bases and columns represent 0:16 incorrect bases.
For example the first plots shows that for T-bases of length 1
surrounded by bases on left and right that are not T, you are most
likely to observe a single correct T base and no other bases. There is
some chance you miss the base or have an extra correct base (above and
below the highest), but you still don't observe many incorrect bases
(partly due to the way I tally).
Note G's have more variance in general.
Colored plots replace missing data (there are no runs of C 5 or
greater for this sample).
These error matrices can be used to detect minor variants and mitigate
noisy haplotypes using CRFs and linear system theory.
T G C A
1 


2 
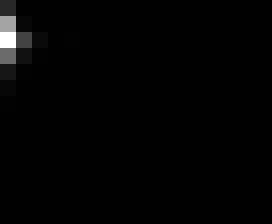

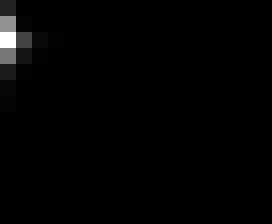
3 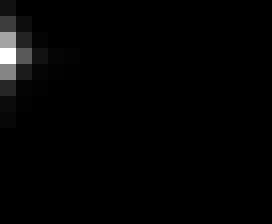
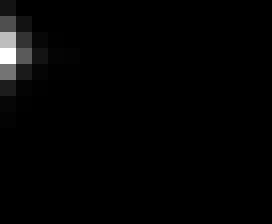
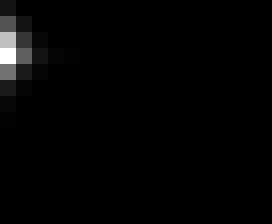
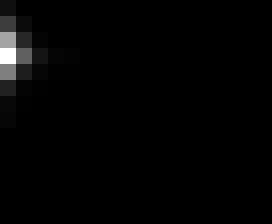
4 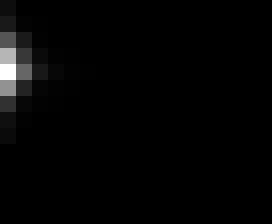
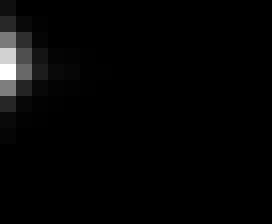
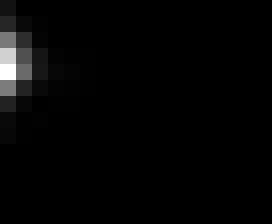
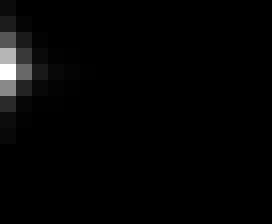
5 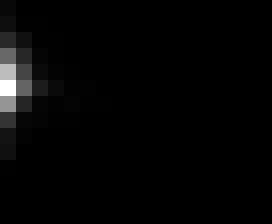
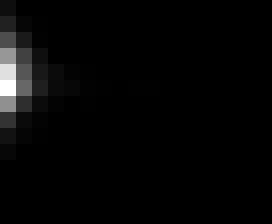

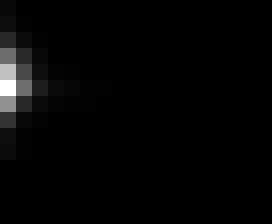
6 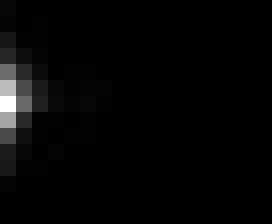
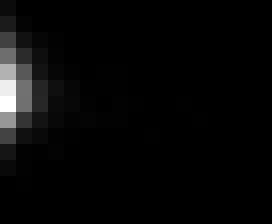
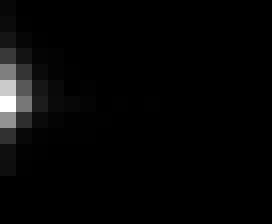
7