WashU BCR-ABL Kinase Domain Sequencing. June 2013
- Goal: sequence two BCR-ABL kinase domain samples from WashU.
- Nested PCR to yield ~875-bp amplicons.
- Samples: one entry (12-360a) and one failure (12-360b: t315i+f359c).
- Does entry show failure variants at low levels?
- Methods: Run codon aware bcr-abl analysis and compare to SMRTporal
compound variant workflow
Next
================================
 Next
================================
Next
================================
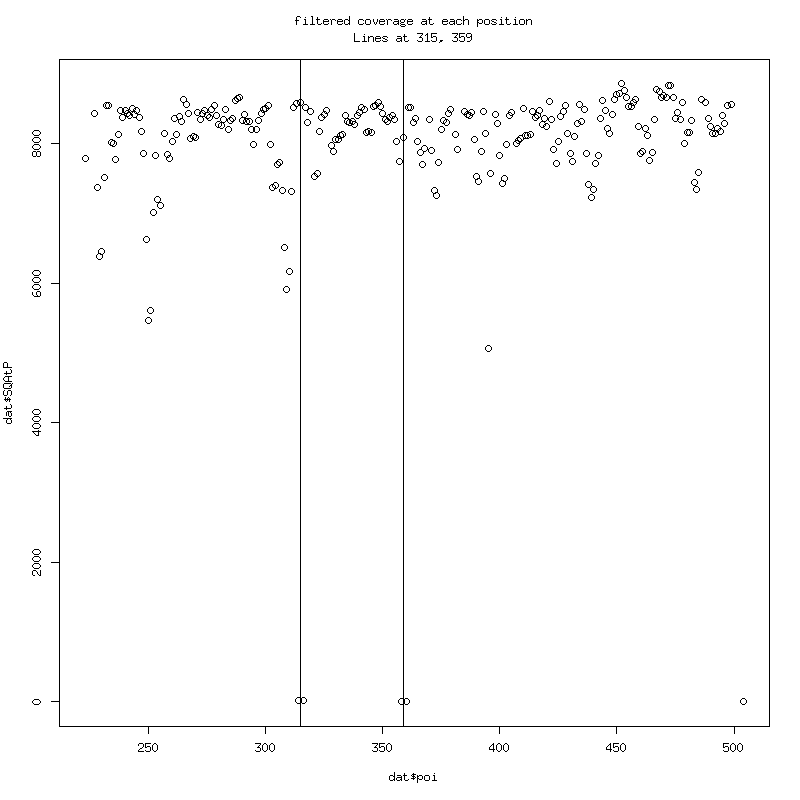
 Note the almost zero coverage to left and right of 315 and 359 due to
wildtype filtering.
Next
================================
Note the almost zero coverage to left and right of 315 and 359 due to
wildtype filtering.
Next
================================
Variant Results
- Entry Sample: - f359c (ttc->tgc) present at nearly 100%. - No evidence of t315i (act->att) (4 out of 10654 reads, less than expected noise). - Possible d241h, k404r, l387l, p408l, and l364l above 1% abundance. - Failure Sample: - Both f359c and t315i present at nearly 100%. - h396y (cat->tat) variant at 32% (2454 / 7566 reads). - Possible w235*, k419e, t495a, and n322n above 1% abundance. - Standard compound variant analysis in SMRTportal finds the same variant positions. - Both samples have what appears to be a ~300bp exon deletion in 0.1% of the reads. Next ================================Analysis Methods
- Basis of variant detection analysis: - align reads to a reference - estimate whether minor variant counts in independent columns: - are due to noise or - have excess of counts due to the presence of a true minor variant - With high enough coverage, this detection can be made to arbitrarily low abundances. - Codon-aware analysis filters away any non-3-base codon position mitigating indel errors. Additional wildtype enforcement in local neighborhood is made. - SMRTPortal Minor and Compound Variants protocol is not codon aware and makes per-base variant calls. Next ================================120_360A Entry Analysis
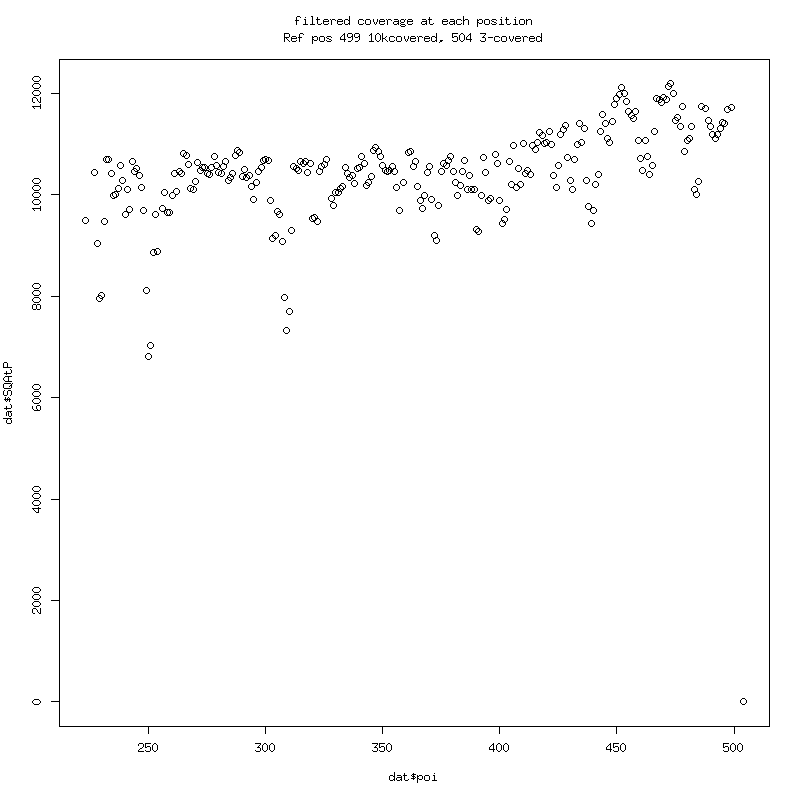
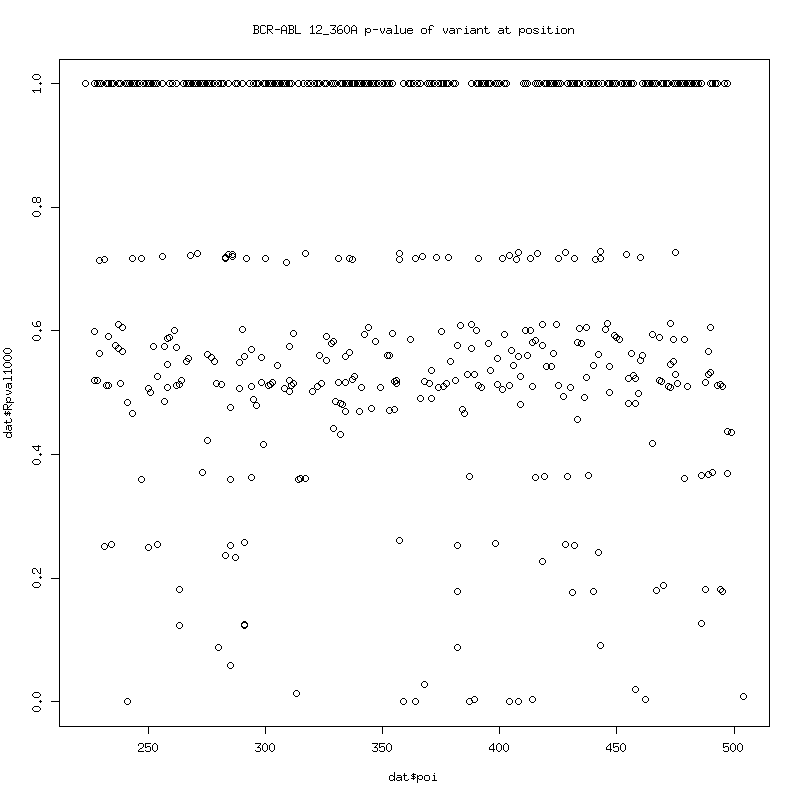
Filtered Coverage: P-values of variant detection at each position:
Next
================================
120_360A Entry Variants
poi coi Qcodon fracNull countNull SNull fracQAtP countQAtP SQAtP mutation Rpval1000
--- --- ------ -------- --------- ----- -------- --------- ----- -------- ---------
359 ttc tgc 0.00022 4 18461 0.99678 10212 10245 f359c 4.674942e-288
241 gac aac 0.00034 14 41057 0.05316 537 10102 d241n 3.543524e-20
404 aaa aga 0.00013 5 38483 0.03982 424 10648 k404r 4.999283e-15
387 ttg ctg 0.00022 4 18064 0.03398 353 10389 l387l 9.988930e-13
408 ccc ctc 0.00027 10 37527 0.02179 229 10511 p408l 5.638817e-08
364 ctt ctc 0.00117 45 38608 0.01042 111 10653 l364l 9.561592e-04
462 gaa gag 0.00026 10 38891 0.00913 101 11068 e462e 3.086022e-03
414 aac gac 0.00010 1 10000 0.00865 95 10980 n414d 4.464454e-03
389 aca gca 0.00014 5 36048 0.00891 90 10103 t389a 4.548214e-03
313 atc acc 0.00010 1 10000 0.00751 79 10522 i313t 1.414940e-02
458 atg gtg 0.00010 2 19878 0.00670 78 11649 m458v 2.008483e-02
368 aac agc 0.00013 5 37254 0.00641 64 9982 n368s 2.821238e-02
Variants with p-value<5%.
f359c is almost universally present.
d241n at 5%. Others might be real but a correct null negative control
run should be done to normalize statistics.
Next
================================
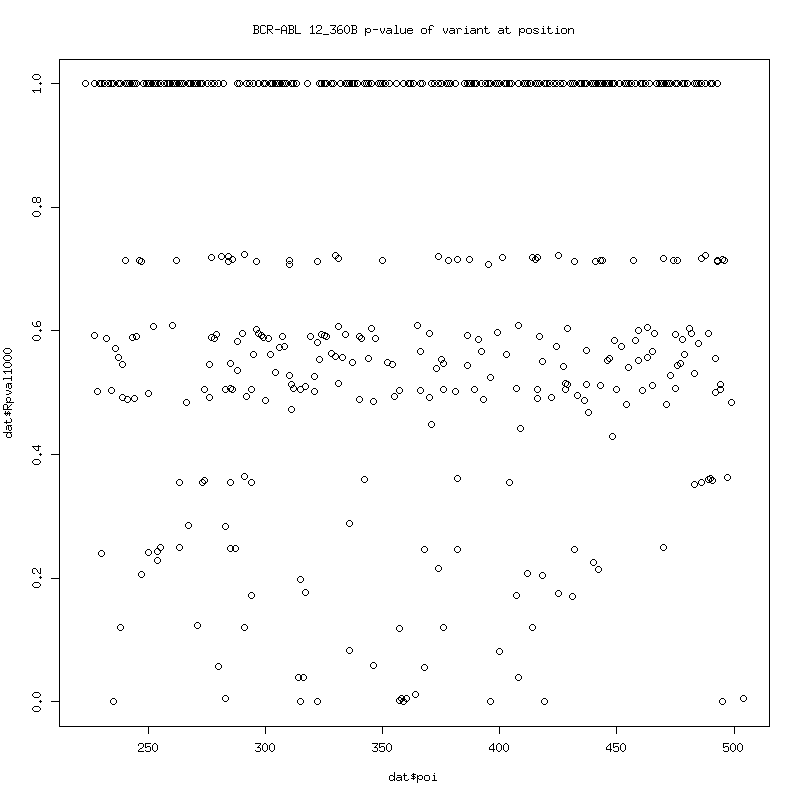
120_360B Failure Analysis
Filtered Coverage: P-values of variant detection at each position:
Note the almost zero coverage to left and right of 315 and 359 due to
wildtype filtering.
Next
================================
120_360B Failure Variants
poi coi Qcodon fracNull countNull SNull fracQAtP countQAtP SQAtP mutation Rpval1000
--- --- ------ -------- --------- ----- -------- --------- ----- -------- ---------
315 act att 0.00392 164 41835 0.99453 8546 8593 t315i 7.240561e-286
359 ttc tgc 0.00022 4 18461 0.99728 8066 8088 f359c 9.054592e-286
396 cat tat 0.00019 7 36645 0.32435 2454 7566 h396y 9.681266e-114
235 tgg tag 0.00023 9 39566 0.03449 276 8002 w235* 8.012982e-13
419 aag gag 0.00016 3 18209 0.02333 195 8360 k419e 1.123321e-08
495 aca gca 0.00027 11 40979 0.01810 152 8396 t495a 1.511495e-06
322 aac aat 0.00015 3 20300 0.01428 108 7565 n322n 5.188290e-05
357 aaa aga 0.00018 7 38728 0.00969 75 7742 k357r 1.739895e-03
283 ttc ctc 0.00010 1 10000 0.00837 71 8487 f283l 5.882372e-03
364 ctt cct 0.00013 5 38608 0.00742 62 8359 l364p 1.253980e-02
408 ccc cct 0.00021 8 37527 0.00621 50 8049 p408p 3.964793e-02
Variants with p-value<5%.
t315i, f359c is almost universally present. h396y at 32.435%.
stop w235* at 3.449%. Others might be real but a correct null negative
control run should be done to normalize statistics.
Next
================================
h396 Drill Down
Show the unfiltered alignments for the 3 codons centered around the 32% h396y position.
count rawReadIdentity
4747 G....C....C....C....A....T....G....C....T....G
2292 G....C....C....T....A....T....G....C....T....G
383 G....C....C....-....A....T....G....C....T....G
352 G....-....C....C....A....T....G....C....T....G
258 G....C....C....Ct...A....T....G....C....T....G
30 G....C....Ct...C....A....T....G....C....T....G
28 G....C....C....C....A....T....G....C....T....-
15 Gg...C....C....C....A....T....G....C....T....G
...
Clear top two identities (GCC.CAT.GCT and GCC.TAT.GCT) showing the 32%
cat->tat variant.
False insertion / deletion artifacts occur at lower frequency and are
filtered by codon-aware analysis.
Insertion/deletions errors coupled with alignment artifacts can lead
to loss of information. We are working on sophisticated analysis
techniques to mitigate this in pricipled ways.
Next
================================
SMRTPortal Comparison
The data was run through the standard SMRTPoral Minor and Compound Variants protocol. 12_360A:
Pos Variant Type Freq Cov Conf (AA) (frame012)
418 418T>G SUB 10795 10948 93 359 1
63 63G>A SUB 534 10903 93 241 0
553 553A>G SUB 432 11294 93 404 1
501 501T>C SUB 351 11100 93 387 0
501 501T>C SUB 351 11100 93 387 0
565 565C>T SUB 225 11336 93 408 1
12-360B:
Pos Variant Type Freq Cov Conf (AA) (frame012)
286 286C>T SUB 8581 8709 93 315 1
418 418T>G SUB 8572 8668 93 359 1
528 528C>T SUB 2453 8716 93 396 0
46 46G>A SUB 278 8744 93 235 1
597 597A>G SUB 202 8803 93 419 0
825 825A>G SUB 147 8912 82 495 0
308 308C>T SUB 121 8696 37 322 2
Looks good. SMRTPortal finds the exact SNP variants that are part of
the codon analysis (cutting at 1% abundance)
Compute aa, frame: aa=((refPos-1-2)/3)+221, frame=(refPos-3)%%3
(account for 0- or 1- based, reference frame, canonical position).
Reference: bcr-abl.reference.fasta
Next
================================
Large Deletion
There are reads with large exon deletions > 300bases to the reference. About 0.1% of reads show deletion but are easily detectable because all variants are contiguous and anchored on both sides. Here is the alignment of all large deletion reads in 12-360B with the reference at the top (best viewed in Firefox or other browser that shows long lines): 12_360B_clucon.largedelete.msa Long reads allow us to see this deletion easily. Next ================================Compound variant analysis
Examine compound variants by looking at fully-spanning reads and counting number of reads that contain different combinations of the variants. Some complications arise because of ambiguous codons (ie not 3 bases in sequencing), these "bleed" counts over a larger number of combinations. Below are two tables representing the compound variant counts. The top #-part lists all of the mutations that are tracked. This is followed by lines listing: counts, fractions, and compound mutants. Ambiguous codons are marked by xxx. For example:
2063 0.240331 t315i.att,f359c.tgc
440 0.051258 t315i.att,f359c.tgc,x396x.xxx
The first line says 2063 reads (24% of the total) contain both t315i
and f359c variants and no other variants (all exactly wildtype)
The second line says 440 reads (5% of the total) contain t315i+f359c
but are ambiguous at 396 with all other positions being wildtype.
12_360A: control_codonMutAnalysisALLPOS-bcr-abl-12_360A.compounds.850.txt
12_360B: control_codonMutAnalysisALLPOS-bcr-abl-12_360B.compounds.850.txt
----
Here is an example using this data. Looking at 12_360B variants: h396y
(32%) and k419e (2.3%). Do these occur together or are they
independently varying?
Our first hint is from the count of the most abundant mutation pattern
that contains k419e (toward the bottom of the result file):
70 0.008155 t315i.att,f359c.tgc,h396y.tat,k419e.gag
The most abundant observation of k419e is compounded with t315i+f359c
(not surprising because they occur at nearly 100%) and h396y (somewhat
surprising because it only occurs 32% of the time, so maybe it's
compounded).
To quantify this, we can count how many times the two variants occur
together and by themselves giving a 2x2 table. (I simply eliminated
any patterns that contained ambiguities "xxx" for this.)
419yes 419no
396yes 80 1101
396no 13 2309
Then we estimate what the counts would look like if both were
independently varying and see if the observed counts are different
(using chi-squared test). This gives me a p-value <2.2e-16, so they
are not independently varying.
Essentially, if variant 419 is present then most likely variant 396 is
compounded with it (80 counts), versus 396 not being there (13
counts).
A more sophisticated analysis of compounds can also be done.
Next
================================