Initial results NIAID reservoir full HIV genomes 2013 October 21st
- GOAL: examine full length HIV genomes isolated from reservoir to gather evidence of whether the viruses are viable - A number of samples were sequenced and analyzed with the goal of obtaining continuous long-reads that cover the entire HIV genome - Summary: - We obtained full length HIV genomes with good yield for most of the samples. - Two genomes were determined to be hyper-mutated. - Runs were single genome samples except for Pt1#1 which is a complex mixture. - Pt1#1 is a mixture of 30 genomes by preliminary estimates. There is a large contiguous-deletion variant subpopulation. - A control experiment of a 90%/10% clone mix was successfully separated into constituent genomes. Next ================================Inputs and Methods
- I used the HXB2 HIV reference as an initial generic reference. After examining coverage, I trimmed the reference to the highly covered bases: 700:9620 (length=8921). Here is the trimmed reference: hiv_hxb2_whole_genome-covered.fasta - Here is the run table of examined runs (two clones, 9 patients, two clone mixes):
runNumber sampleName
2450618-0001 pDH12
2450618-0002 pNL43
2450618-0003 Pt1#1
2450618-0004 Pt2#1
2450618-0005 Pt3#1
2450618-0006 Pt4-1#1
2450618-0008 Pt4-2#1
2450618-0007 Pt5#1
2450618-0009 Pt6#1
2450618-0010 Pt7#1
2450618-0011 Pt8#1
2450618-0012 1%pNL43
2450618-0013 10%pNL43
- More runs were done but are not analyzed in this document.
- ClusteringConsensus is used to estimate a per-sample consensus
reference, align raw reads to that reference, and keep only those
that are fully-spanned to within 1%. Multiple alignments are
generated and reads in the alignments are clustered to look for any
structure. Various statistics are reported.
- Hyper-mutations were examined using the tool at www.hiv.lanl.gov.
Next
================================
Mapping statistics
#### total number of reads
raw-Pt5#1.fasta 316 x
raw-Pt8#1.fasta 3456 x
raw-Pt3#1.fasta 6622 x
raw-Pt4-1#1.fasta 25081
raw-1%pNL43.fasta 29811
raw-Pt7#1.fasta 45496
raw-Pt6#1.fasta 46110
raw-Pt2#1.fasta 48233
raw-Pt4-2#1.fasta 50126
raw-pDH12.fasta 56190
raw-pNL43.fasta 63413
raw-Pt1#1.fasta 66180
Three runs have low number of reads. Three were noted to have very low
loading.
#### number of mapped reads
249 clucon-Pt5#1/alignments.cmp.h5.error x
1336 clucon-Pt8#1/alignments.cmp.h5.error x
2347 clucon-Pt3#1/alignments.cmp.h5.error x
23450 clucon-Pt2#1/alignments.cmp.h5.error
23925 clucon-Pt4-1#1/alignments.cmp.h5.error
28826 clucon-1%pNL43/alignments.cmp.h5.error
40375 clucon-Pt7#1/alignments.cmp.h5.error
41564 clucon-Pt6#1/alignments.cmp.h5.error
44551 clucon-Pt4-2#1/alignments.cmp.h5.error
53896 clucon-pDH12/alignments.cmp.h5.error
56072 clucon-pNL43/alignments.cmp.h5.error
57814 clucon-Pt1#1/alignments.cmp.h5.error
#### number of full length (99%) mapped reads
3 clucon-Pt5#1/alignments.filterFull x
55 clucon-Pt8#1/alignments.filterFull x
192 clucon-Pt3#1/alignments.filterFull x
645 clucon-Pt2#1/alignments.filterFull
1618 clucon-pNL43/alignments.filterFull
1660 clucon-1%pNL43/alignments.filterFull
1668 clucon-Pt7#1/alignments.filterFull
1857 clucon-pDH12/alignments.filterFull
1950 clucon-Pt6#1/alignments.filterFull
2006 clucon-Pt4-2#1/alignments.filterFull
2401 clucon-Pt1#1/alignments.filterFull
2427 clucon-Pt4-1#1/alignments.filterFull
Next
================================
Consensus Genomes
- Give single consensus genome for each sample:
Consensus Length
clucon-Pt3#1/quiverResult.consensus.fasta 8456
clucon-Pt6#1/quiverResult.consensus.fasta 8792
clucon-Pt5#1/quiverResult.consensus.fasta 8867
clucon-1%pNL43/quiverResult.consensus.fasta 8898
clucon-pDH12/quiverResult.consensus.fasta 8899
clucon-pNL43/quiverResult.consensus.fasta 8906
clucon-Pt4-2#1/quiverResult.consensus.fasta 8922
clucon-Pt1#1/quiverResult.consensus.fasta 8924
clucon-Pt4-1#1/quiverResult.consensus.fasta 8924
clucon-Pt2#1/quiverResult.consensus.fasta 8960
clucon-Pt8#1/quiverResult.consensus.fasta 8986
clucon-Pt7#1/quiverResult.consensus.fasta 8988
Fastq:
clucon-Pt3#1/quiverResult.consensus.fastq
clucon-Pt6#1/quiverResult.consensus.fastq
clucon-Pt5#1/quiverResult.consensus.fastq
clucon-1%pNL43/quiverResult.consensus.fastq
clucon-pDH12/quiverResult.consensus.fastq
clucon-pNL43/quiverResult.consensus.fastq
clucon-Pt4-2#1/quiverResult.consensus.fastq
clucon-Pt1#1/quiverResult.consensus.fastq
clucon-Pt4-1#1/quiverResult.consensus.fastq
clucon-Pt2#1/quiverResult.consensus.fastq
clucon-Pt8#1/quiverResult.consensus.fastq
clucon-Pt7#1/quiverResult.consensus.fastq
Next
================================
Variant Positions
- Number of positions that are likely to contain minor variants according to simple entropy threshold in the multiple alignments
Num Sample
0 clucon-Pt4-1#1
0 clucon-1%pNL43
0 clucon-pDH12
1 clucon-Pt2#1
2 clucon-Pt6#1
4 clucon-pNL43
5 clucon-Pt7#1
6 clucon-Pt8#1 x
6 clucon-Pt4-2#1
8 clucon-Pt3#1 x
91 clucon-Pt1#1
1209 clucon-Pt5#1 x
- Only Pt1#1 appears to have a good number of variant positions. The
others might be to untuned thresholds. (x discounts the low coverage
runs)
Next
================================
Clustering
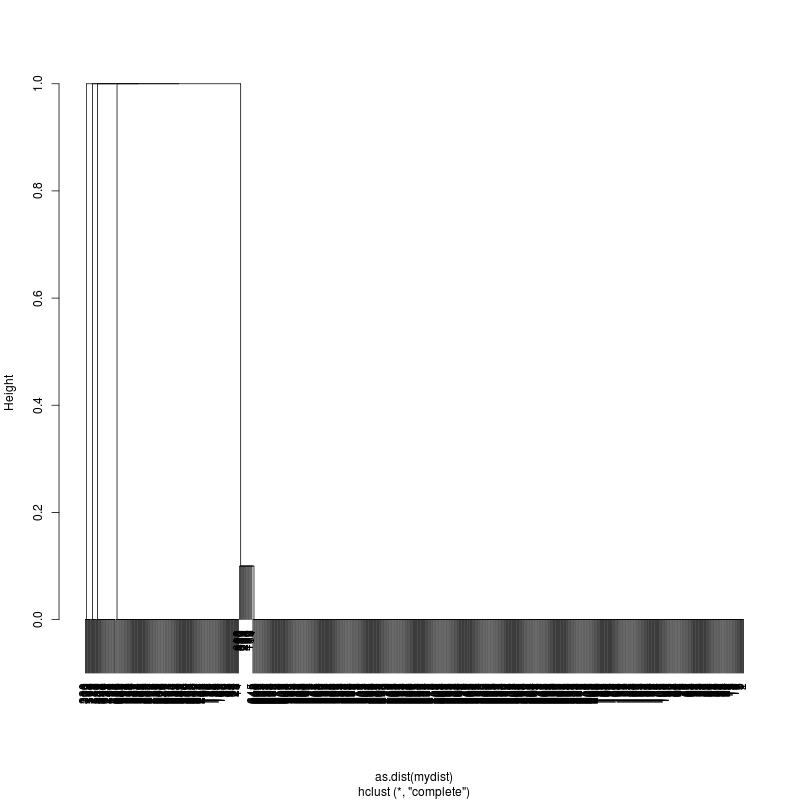
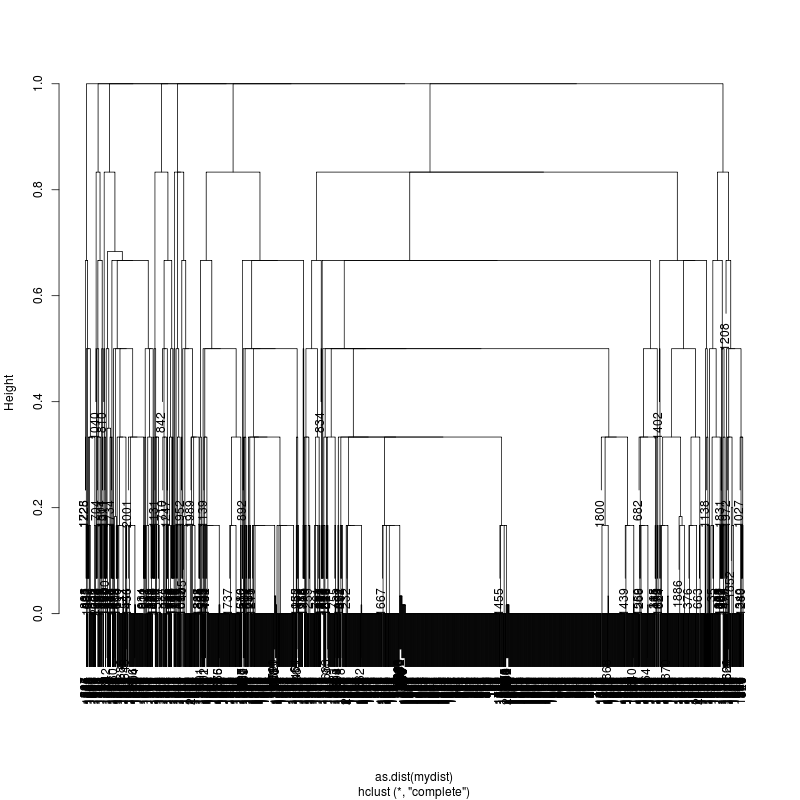
- Examine the complete-linkage clustering of all reads on variant positions. Pt1 Pt2 Pt3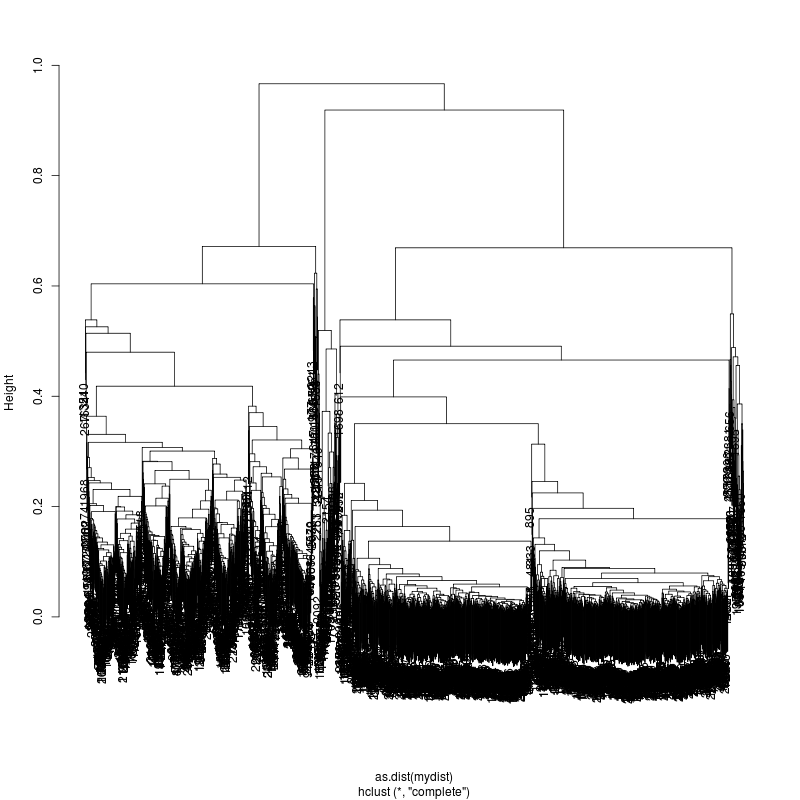
 Pt4-2 Pt5 Pt6
Pt4-2 Pt5 Pt6
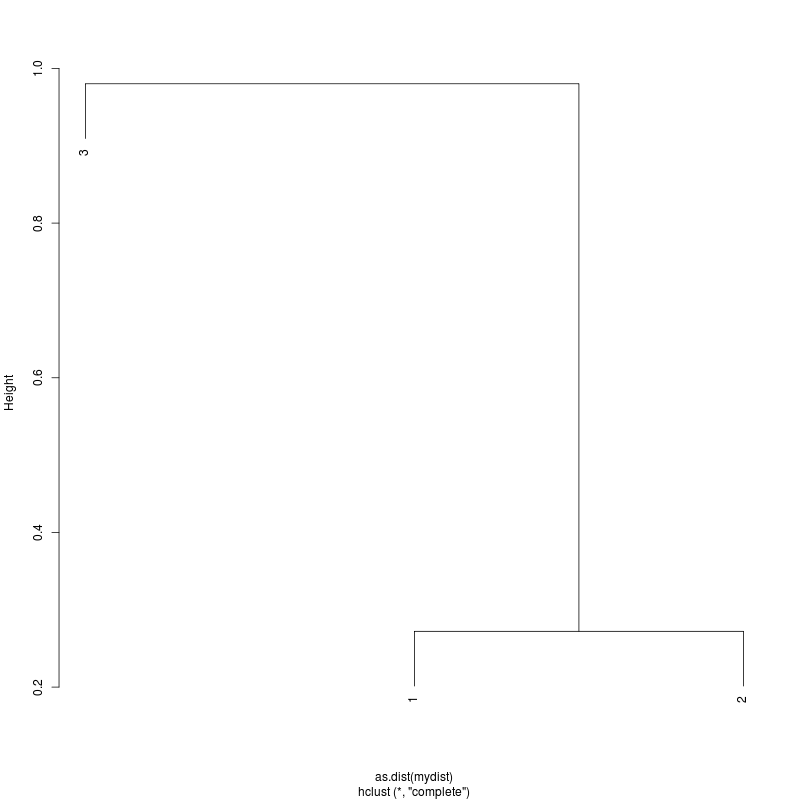
 Pt7 Pt8 pNL43
Pt7 Pt8 pNL43
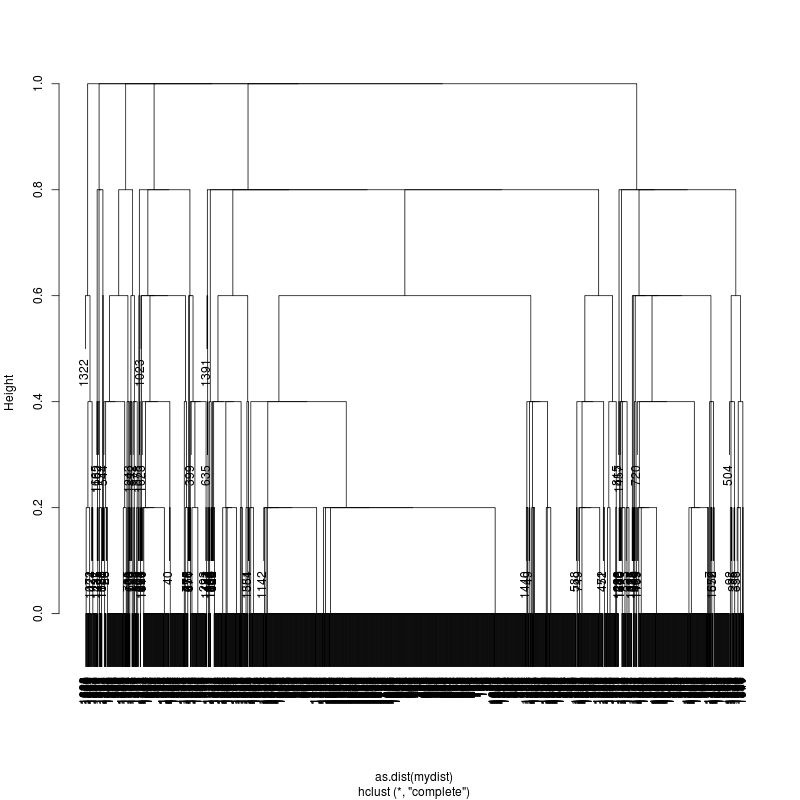
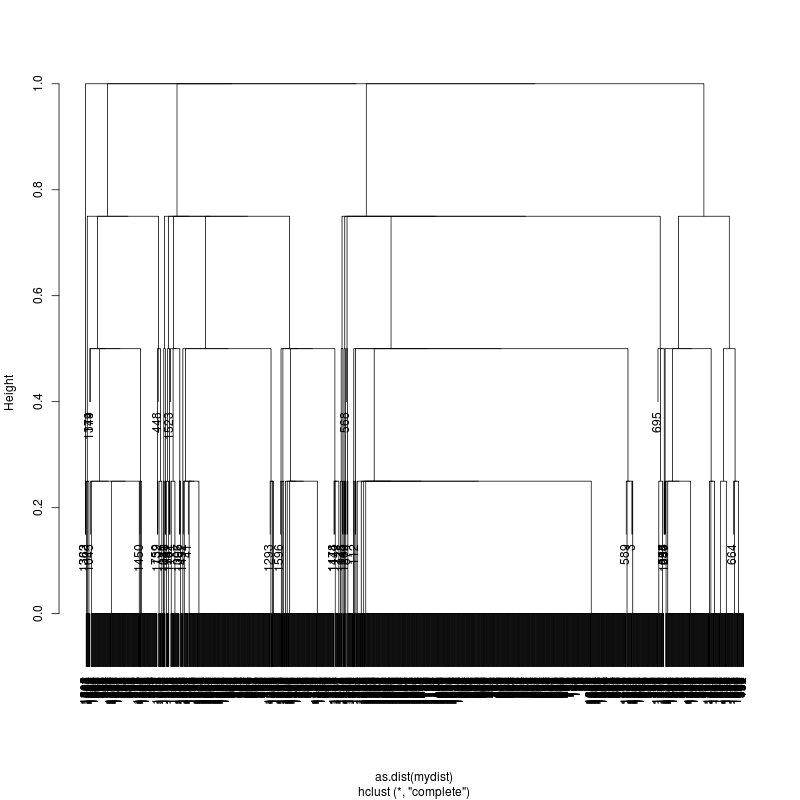 - Note missing plots had no variant positions on which to cluster.
- Each column is a full-length amplicon-spanning read and the y-axis
represents the distance which is the fraction of variant positions
that disagree (0=identical, 1=completely different over the variant
positions). For example, a join distance of 0.8 between subclusters
says that every pairwise distance in the subtree is less than 0.8.
- Only Pt1#1 has complexity and likely multiple genomes.
Next
================================
- Note missing plots had no variant positions on which to cluster.
- Each column is a full-length amplicon-spanning read and the y-axis
represents the distance which is the fraction of variant positions
that disagree (0=identical, 1=completely different over the variant
positions). For example, a join distance of 0.8 between subclusters
says that every pairwise distance in the subtree is less than 0.8.
- Only Pt1#1 has complexity and likely multiple genomes.
Next
================================
Truth Comparison
- We were given Sanger reference sequences for NL43, DH12, Pt1, and Pt4-2. - Show alignments of PacBio consensus to Sanger (Sanger top, PacBio bottom) (Best viewed in long line browser to see alignment, Firefox) - NL43: blasr.pNL43.CMRS.output (4 G, 1 C homopolymer deletions) - DH12: blasr.pDH12.CMRS.output (8 G, 1 C homopolymer deletions) - Pt1: (heterogeneous sample) blasr.Pt1.output ( many differences with large delete ) - Pt4-2: blasr.Pt4-2.output (1 G, 4 A, 1 C homopolymer deletions) - Note several homopolymer deletion errors. This is caused by a software bug that didn't allow full base sequencing information (QVs) to be used in consensus. This is being fixed and should lead to elimination of the homopolymer deletion errors. Next ================================Hyper-mutation Analysis
- Hyper-mutations are estimated by aligning the consensus to HXB2 and submitting the two sequence alignment to http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html
Sample Hypermutation p-value counts
1%pNL43 95.5%
pDH12 94.1%
pNL43 83.0%
Pt1#1 68.1%
Pt2#1 87.1%
Pt3#1 83.7% X
Pt4-1#1 1.11e-53 334/1109
Pt4-2#1 6.73e-52 336/1105
Pt5#1 70.0% X
Pt6#1 91.6%
Pt7#1 95.5%
Pt8#1 98.7% X
X marks low yield runs
- Pt4-1#1 and Pt4-2#1 are hyper-mutated
Next
================================
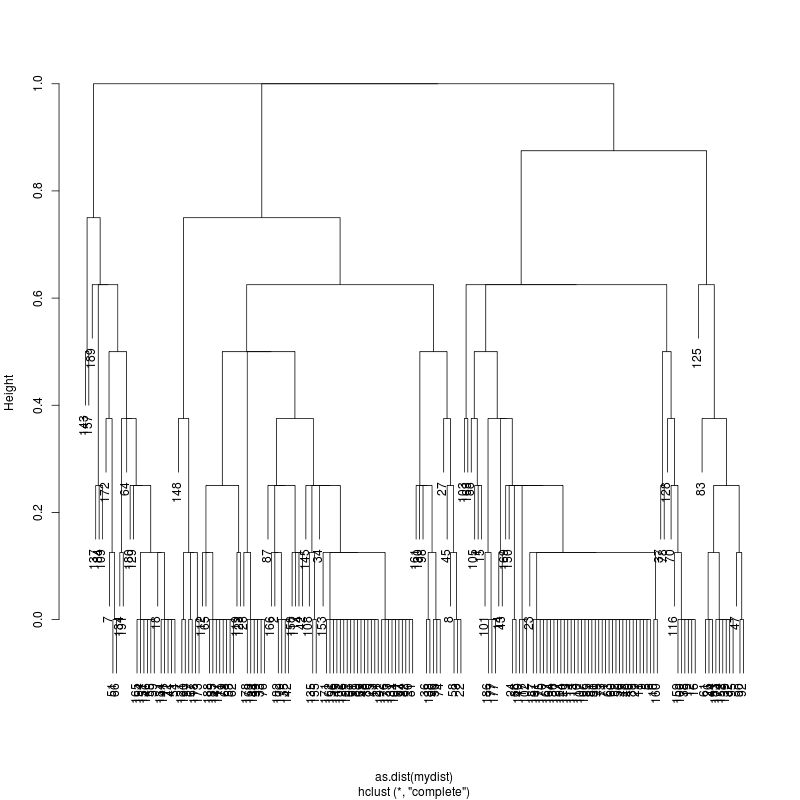
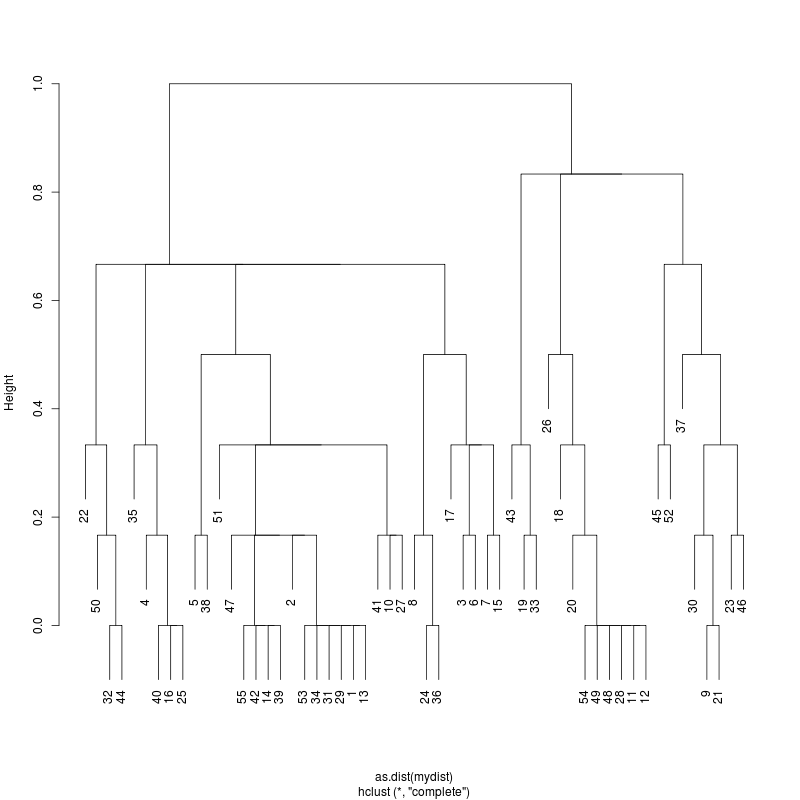
Pt1 Mixture
- Pt1 sample was estimated to be a complex mixture.
- Run ClusteringConsensus recursively until all reads in subgroup are
determined to be single genome.
- Recursive Tree:
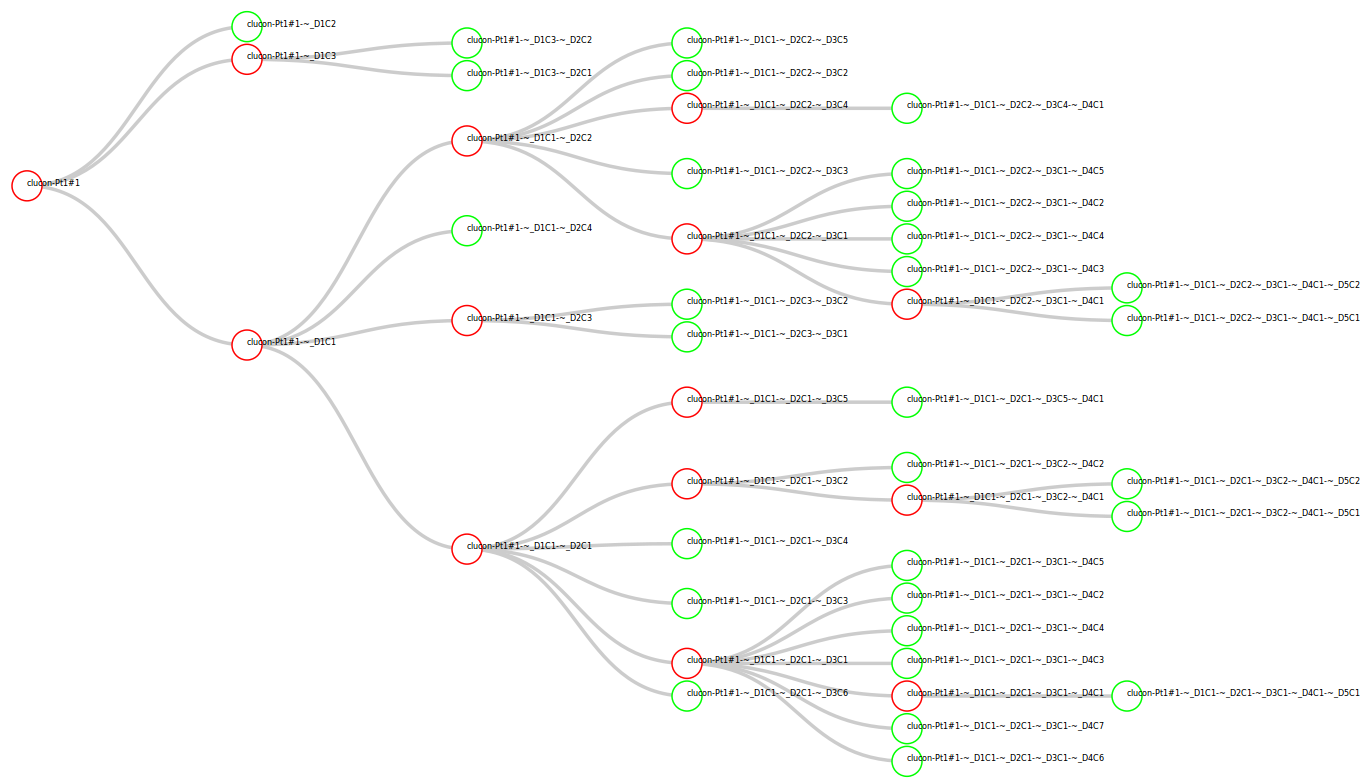 Next
================================
Next
================================
Pt1 Deconvoluted Consensus
- For each single genome child give consensus genome
SubspeciesConsensus ConsensusLength
clucon-Pt1#1-~_D1C2 8873
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C1-~_D5C1 7371
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C1-~_D5C1 7370
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C2-~_D4C1-~_D5C1 7367
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C4-~_D4C1 7366
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C2 7366
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C3 7365
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C7 7363
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C3 7362
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C2 7361
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C5-~_D4C1 7361
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C5 7360
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C2-~_D4C1-~_D5C2 7359
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C3 7359
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C6 7359
clucon-Pt1#1-~_D1C1-~_D2C3-~_D3C1 7358
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C3 7357
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C4 7357
clucon-Pt1#1-~_D1C1-~_D2C4 7357
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C2 7355
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C1-~_D5C2 7355
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C4 7355
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C1-~_D4C6 7348
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C2-~_D4C2 7347
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C5 7345
clucon-Pt1#1-~_D1C3-~_D2C2 7343
clucon-Pt1#1-~_D1C1-~_D2C1-~_D3C4 7342
clucon-Pt1#1-~_D1C1-~_D2C2-~_D3C1-~_D4C5 7333
clucon-Pt1#1-~_D1C3-~_D2C1 7316
clucon-Pt1#1-~_D1C1-~_D2C3-~_D3C2 7239
- Largest genome is 8873 bases and next largest is 7371 bases, a large
deletion variant.
- Note: this is an initial result. The algorithm is complex and has
tunable parameters that must be further optimized. Consequently, our
confidence in the 30 genomes is not as high as it would be after
tuning.
Next
================================
Large Deletions
- Pt1 has large-deletion subspecies. ( This is corroborated by a second band seen in the sizing gel ) - Alignment of the two longest consensus genomes (8873 bases vs 7371 bases) pt1sub2longest.blasr A large contiguous deletion! - This deletion variant can be seen using simple methods: - 1. Plot cumulative sum of deletions for each read in multiple alignment (contiguous deletions go up at angle). 2. Plot density of raw mapped readlengths.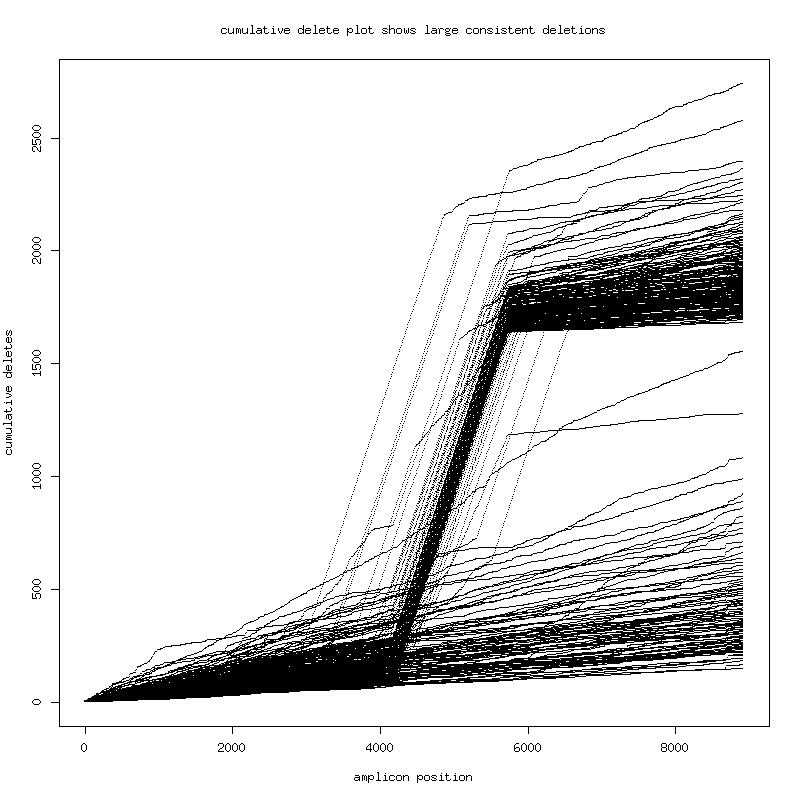
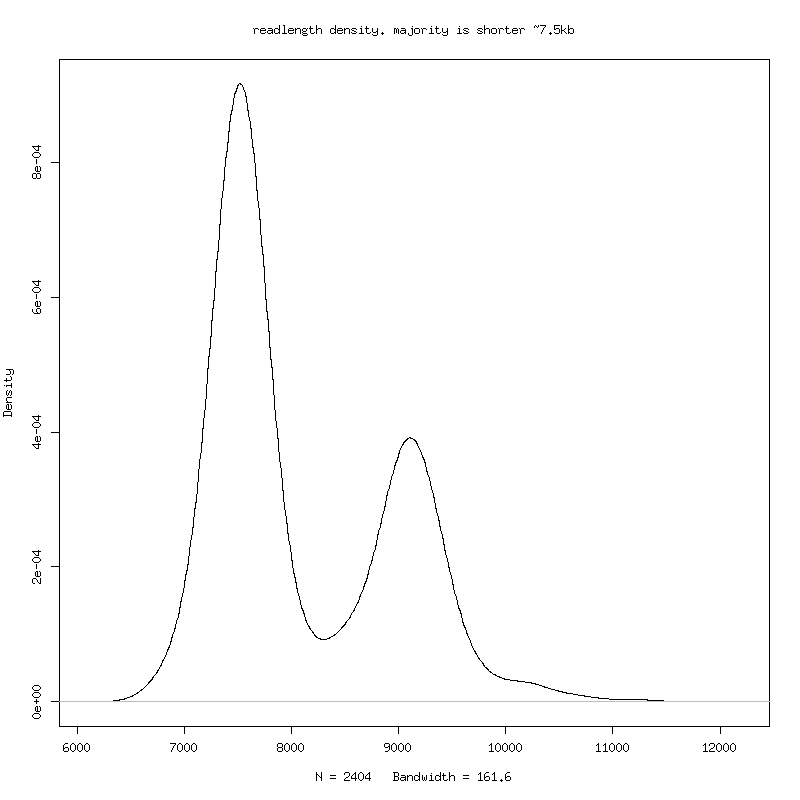 There is a population of ~1750 base deletion (4000:5750 in the
sample consensus reference).
Next
================================
There is a population of ~1750 base deletion (4000:5750 in the
sample consensus reference).
Next
================================
Control clone mixture
- For a positive control for mixture deconvolution, sequence a mixture of clones: 90% pDH12 / 10% pNL43. - ClusteringConsensus starts with generic HXB2 and estimates mixture consensus. It then discovers 95 variable positions and clusters. (Alignment of Sanger reads shows 725 differences. Lower coverage might cause lower number to be discovered ) - Clustering plot on those 95 positions show minor subspecies: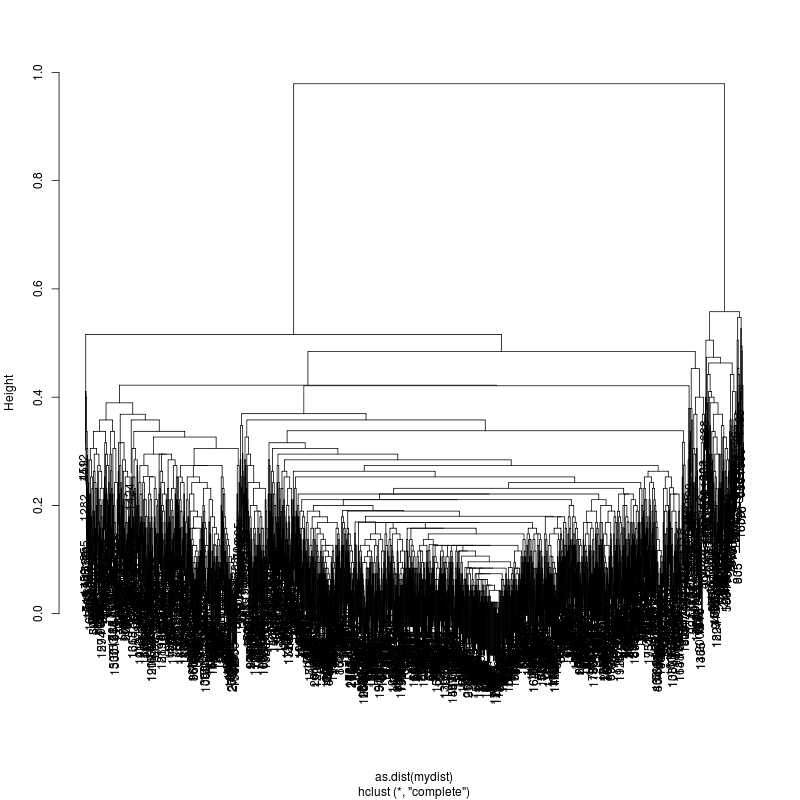 - Stratifying reads into those two clusters shows 117 reads in minor
and 1907 reads in major. This is a 5.8% minor mixture which might
just be an imperfect mix of the 10% designed.
- Aligning the two consensus sequences from the deconvolution to
Sanger truth shows:
- DH12 (20 homopolymer deletions only)
10percent-dh12-D1C1.blasr
- NL43 (9 homopolymer deletions only)
10percent-nl43-D1C2.blasr
- NOTE: software bug not using full sequencing information and lower
coverage from stratification cause more HPdel errors. With
software fix, we expect most to all of these errors to disappear.
- Knowing nothing more than the sample contains HIV, we are able to
extract two complete HIV genomes and estimate the mixture proportion.
Next
================================
- Stratifying reads into those two clusters shows 117 reads in minor
and 1907 reads in major. This is a 5.8% minor mixture which might
just be an imperfect mix of the 10% designed.
- Aligning the two consensus sequences from the deconvolution to
Sanger truth shows:
- DH12 (20 homopolymer deletions only)
10percent-dh12-D1C1.blasr
- NL43 (9 homopolymer deletions only)
10percent-nl43-D1C2.blasr
- NOTE: software bug not using full sequencing information and lower
coverage from stratification cause more HPdel errors. With
software fix, we expect most to all of these errors to disappear.
- Knowing nothing more than the sample contains HIV, we are able to
extract two complete HIV genomes and estimate the mixture proportion.
Next
================================